
Introduction
ANVISA, the Brazilian human health regulatory authority, has issued in recent years a number of documents describing in detail what is expected to be presented in a Regulatory submission in Brazil for Forced Degradation Studies (FDS), i.e., how FDS should be designed and managed, and how relevant data should be evaluated (Ref. 1-3).
The target of this blog is to summarize at high level, in an easy and schematic way, following the FDS process flow, the key elements to be considered when designing and assessing data from an FDS intended for ANVISA submission.
Part 1 of this blog (Ref. 4) covered background and basic information, both on FDS according to ICH and WHO stability guidelines and on ANVISA guidance, while Part 2 (Ref. 5) covered how ANVISA considers FDS in an overall context of a Degradation Profile Study, how to prepare an FDS protocol, and how to choose the right batch of drug substance or drug product to be tested in the FDS.
The present third and final part of the blog will mainly cover how to choose the stress storage conditions, how to design and when to terminate the study, and finally, how to prepare the overall report according to ANVISA requirements and recommendations.
Choice of FDS stress storage conditions according to ANVISA
ANVISA’s general requirements for storage conditions to be applied for an FDS on the drug substance (API) are reported in the RDC 318/2019 (Ref. 6 – Art. 37). Required storage conditions for the API FDS study are temperature, humidity, oxidation, light, and susceptibility to hydrolysis over a wide range of pH values (Ref. 6 – Art. 37). A technical justification must be included in the report if omitting a required condition, e.g., if any condition cannot be applied, due to the inherent characteristics of the sample, or if it is generally not applicable (Ref. 6 – Art. 37).
If an FDS of a drug substance is being performed in the context of, or in preparation for an FDS for a drug product, specific requirements for drug product FDS, as listed below, should also be considered.
All the above mentioned ANVISA-required FDS conditions are quite similar to the ICH Q1A (R2) conditions recommended for an API FDS (Ref. 7 – Sect. 2.1.2.).
General requirements by ANVISA for storage conditions to be applied for an FDS on a drug product are detailed at a high level in the RDC 53/2015 document (Ref. 1). Additionally, the stability study RDC 318/2019 refers to that same document for designing FDS on a drug product (Ref. 6 – Art. 40).
The RDC 53/2015, related only to the Drug Product, specifically requires that a company should submit FDS data exposing the sample to conditions of heating, humidity, acid solution, basic solution, oxidizing solution, photolytic exposure and metallic ions (Ref. 1 – Art. 5).
It may be useful to underline here the specific new requirement for a drug product FDS with metallic ions, vs. ICH Q1A(R2) recommendations for FDS. This specific ANVISA FDS requirement mainly targets the assessment of potential active oxidative degradation pathways catalyzed by the metallic ions.
A technical justification must be included if not using any one of the above conditions, e.g., if any cannot be applied due to the inherent characteristics of the sample, or if not applicable in general (Ref. 1 – Art. 5).
A more detailed description of how performing the recommended FDS conditions for a drug product for ANVISA is reported in the guideline 04/2015 (Ref. 2 – section 8).
Table 1 below summarizes the ANVISA FDS high level condition requirements included in RDC 53/2015, plus the corresponding detailed recommended conditions reported in the guideline 04/2015.
FDS storage conditions that can be used as a starting point for protocol definition, as recommended in literature for a stable compound (Ref. 9 and 10), are also included in the same table.
Table 1 – ANVISA FDS condition requirements and recommendations vs. typical FDS conditions found in literature
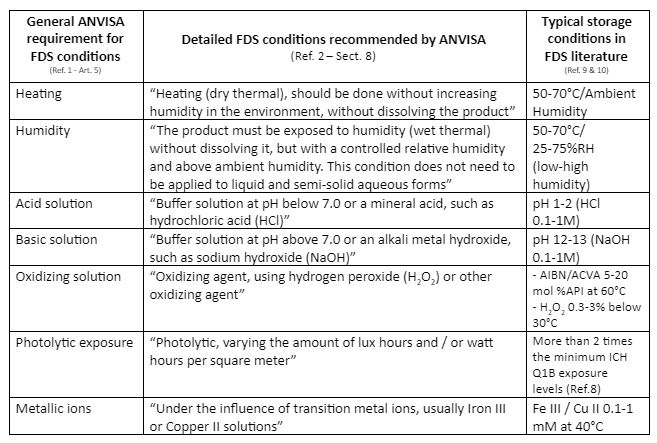
ANVISA requires that all the above-mentioned storage conditions must be equally applied for drug product FDS, for comparison purposes, to the drug substance, to the drug product (all concentrations, unless justified) and to a placebo (Ref. 1 – Art. 4). Furthermore, all the above-mentioned FDS conditions should be applied to all types of formulations and the relevant placebo, unless justified (Ref. 1 – Art. 4).
Let’s expand this ANVISA-required approach, that deviates from current best FDS practices, mainly for solid dosage forms, as indicated in FDS literature (Ref. 9).
According to current practices (e.g., Ref. 9) typically applied FDS conditions to dosage forms are:
- Solid dosage forms: thermal & thermal/humidity, photostability and oxidative (e.g., at solid state under pressurized O2) conditions
- Liquid dosage forms: Acid/base (optional), photostability and thermal conditions
For an FDS intended for ANVISA submission, all the conditions required by ANVISA, including the acid-base and oxidizing solutions and metallic ions (Ref. 11 & 12), as reported in Table 1 above, should also be applied to solid oral dosage forms. Obviously, humidity conditions do not need to be applied to liquid and semi-solid aqueous dosage forms (Ref. 2 – Sect. 8).
This ANVISA requirement for solution FDS as well as for solid drug products, is targeted to accelerate potential reactions between the active and the excipients that would be noticed after long term exposure at normal storage conditions (Ref. 11). This requirement has had a lot of scientific discussion, and as presented by Dr. John Campbell at the May 2021 Science of Stability conference, a benchmarking study was performed involving the regulatory sector and ANVISA. The outcome confirmed that these FDS solution conditions would not provide any additional information vs. a well-designed solid dosage form FDS without a solution study. As soon as the reported study is officially published, the authors of this blog will share the relevant reference within the StabilityHub community, by posting it on the StabilityHub website.
In theory, it could be possible in some instances to waive the FDS experimental exercise for ANVISA following the criteria reported in the guideline 04/2015 (Ref. 2 – Fig. 2 and Ref. 3 – item 4.1.1) or from registered or known degradation profiles for the product (Ref.1 – Art. 8).
However, in practice, it is quite difficult and risky to not have the FDS experimental part when submitting a dossier to ANVISA, especially because the literature does not usually present the criteria established by ANVISA to be able to substitute the results with the API and Drug Product particularities. Additionally, as the degradation profile of a product depends on the API and the adopted excipients, it may be not possible to assess a degradation profile without an FDS experimental part (Ref. 3 – item 4.1.7).
Another ANVISA regulation to consider within an FDS discussion for Brazil is RDC 166/2017 (Ref. 13) that covers the validation of the analytical methods. This regulation also reports ANVISA requirements for showing selectivity of an applied quantitative analytical method (e.g., assay and impurities). Within these requirements a reference is made to performing a supporting FDS.
Table 2 below, summarizes the ANVISA FDS requirements for the manufacturer of an API or a Drug Product, following the indications of RDC 318/2019, RDC 166/2017 and RDC 53/2015.
Table 2 – Comparison of ANVISA FDS requirements for API and Drug Product
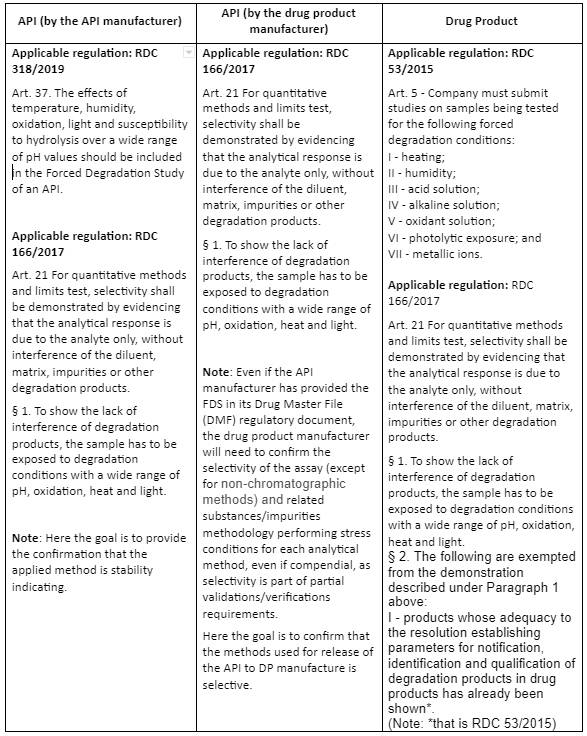
When the RDC 53/2015 report for the drug product is presented (e.g., for a new Marketing Authorizations and applicable variations), it is possible to have all the requirements met for the drug product, that is, conducting the FDS and validating the analytical methodology for the drug product as in the example below:
- Conducting the FDS with the applicable conditions of RDC 53/2015 with the API, drug product, and placebo.
- Evaluating the results with assay and related substances/impurities proposed methodologies using a Reference Standard (SQR) or a Characterized Reference Standard (SQC, i.e., a characterized batch used as per requirements of art. 15 of RDC 166/2017) for standards solutions.
- Assessing peak purity of the API in both assay and drug related impurity analytical methods, if different, and proving that there is no interference in the main peak (Ref. 3 – Item 4.2.19).
- Calculating the mass balance, that is, as defined by ANVISA, the “process of adding the [active] content and levels of degradation products found [at different FDS test points] to assess how the sum approaches 100% of the initial content value, with due consideration of the margin of precision and analytical accuracy.” (Ref.2 – Sect. 4.7).
- Using this data in assessing selectivity of each method (again if different for assay and drug related impurities), referencing RDC 53/2015 report in analytical validation reports.
Active degradation target and study termination at the FDS conditions
It is a specific ANVISA requirement that each FDS condition must produce a degradation greater than 10%, but lower than the one that would lead to complete degradation of the sample, compromising the test (Ref. 1 – Art. 6 – § 1). This requirement is related to the key technical point that an FDS must promote a degradation sufficient to allow evaluation of the formation of degradation products (Ref. 12).
The 10% percent target degradation of initial active content may be achieved by properly defining exposure time, storage temperature, and type and concentration of the degrading agents (Ref. 2 – Sect. 8), as exemplified in the previous section Table 1.
If the achieved degradation is less than 10% at test end at any condition, the company must present a technical justification for this degradation level (Ref.1 – Art. 6 – § 2), confirming that the more drastic conditions were tested.
This last point corresponds to the definition of the so-called study ‘endpoints’. ANVISA has not specifically defined FDS endpoint criteria in its documents and guidelines in addition to the above general indications by ANVISA of minimum target degradation, avoiding an excessive degradation (Ref. 1 – Art. 6 – § 1). This may be especially valid for a compound that may be very stable at a given condition, basically resulting in no or very low degradation by study end. This is due to the lack of agreement or harmonization on this topic in current world-wide regulatory guidelines and literature, as explained by ANVISA itself (Ref. 3 – item 4.2.15).
Therefore, each sponsor must present in the application the references and the justification of the endpoints used, in order to support the FDS (Ref. 3 – Item 4.2.15).
For completeness, there are two main approaches for deciding when an FDS on stable compounds could be ended. The first is based on the rationale that at the end of each stress condition, in absence of significant degradation, an energy at least equivalent to a 6-month storage at 40°C/75%RH (Ref. 11 and 14) should be reached before terminating the study. The other is the WHO position of a max 10-day storage at any condition (Ref. 15) to consider the compound stable at the condition and therefore possibly terminating the study.
It is important to mention that the absence of this justification of the endpoint is a very common point of deficiency letters and rejections from ANVISA.
Study report
As highlighted in Part 2 of this blog series (Ref. 5), ANVISA views an FDS as only the experimental part of a higher level ‘Degradation Profile Study’ (DPS), the critical analysis of a drug substance or drug product degradation profile (Ref. 3 – Item 4.1.1.).
Therefore, ANVISA expectation for an FDS study report is focused on receiving an overall discussion of the product DPS and not just the results of the FDS, which as noted, ANVISA considers to be a supportive experimental tool.
The FDS study report should also include all the key assessments made after the preliminary bibliographic search has been performed and prior to the FDS (as mentioned in Part 2 of this blog series) and how the degradation sensitivity of the compound impacts the manufacturing and storage conditions of the drug product (Ref.2 – Sect. 10).
ANVISA expectations for the content of a FDS/DPS report are detailed in the Guideline 04/2019 (Ref. 2 – Sect. 10). Key requirements can be summarized as:
- Introduction, including highlighting the functional groups of the molecule most susceptible to degradation and the applied method(s) of analysis.
- Experimental Segment, including the FDS information. It should include or describe the:
- Stress exposure parameters (i.e., degrading agent, concentration, temperature and time). If it was not possible to reach a significant active degradation at any of tested stress conditions (i.e., more than 10% of initial), the bibliographic reference used to define the maximum exposure time should be reported
- key chromatograms of each of the API(s), placebo and drug product at each FDS condition with adequate degradation (i.e., more than 10%) or when exposed to the maximum justified exposure time, if not reaching adequate degradation
- result table(s) reporting the FDS results as both degradation percentage of the API(s) with corresponding percentage increase of the different degradation products
- discussion of the study results, also including a comment on the mass balance at all FDS test conditions and a proposal of the degradation mechanisms, if possible. This section should also include discussion of the proposed limits for specified, unspecified and total impurities relating it with notification, identification and qualification limits provided in art. 9 of RDC 53/2015. If any impurity needs to be qualified, this report is submitted in a separate section of the Marketing Authorization (MA) submission.
- Conclusions, including the “potential” degradation profile, with a discussion of both the impact of manufacturing conditions on the degradation profile and of the storage factors that can impact product stability.
The report is presented in section 3.2.P.5.5 (‘Characterisation of Impurities’) when the MA submission or variation is submitted to ANVISA in the Common Technical Document (CTD) format (Ref. 16 and 17), or, as the CTD is not yet mandatory in Brazil, it is presented in item 16 (n) with the other requirements of the development report.
A clarifying whitepaper on the Brazilian CTD and the differences vs. the EU CTD can be found on request in the Vita Regulatory Affairs Consulting website (Ref. 18).
Final Conclusions
A Forced Degradation Study intended to be submitted to ANVISA, to support a marketing authorization application and specific variations in Brazil, although broadly aligned to ICH Q1A (R2), Q1B and WHO stability guidelines recommendations (Ref. 7, 8 and 15), should also consider several points specific for ANVISA, as reported in the present three-part blog. Some key examples are summarized below.
- For the drug product, in addition to those already listed in the ICH Q1A (R2) and Q1B guidelines, adoption of ANVISA’s expressly required stress condition involving challenging with metallic ions, unless justified
- need to achieve a degradation greater than 10% of initial active content at each stress condition, if not properly justified
- application to solid dosage forms of the same FDS conditions applied to drug substance and liquid dosage forms, therefore also including e.g., acid, basic and oxidizing solution conditions
- detailed information to be included in the final FDS report for ANVISA, not only focusing on the FDS data report and comments, but also covering all the main aspects related to the higher level ‘Degradation Profile Study’ required by ANVISA.
As the information included in this three-part blog is intended to supply only high level views on the main ANVISA peculiarities on FDS to the StabilityHub community, the Authors strongly recommend that personnel directly involved in these FDS for Brazil, to review detailed contents of all original ANVISA documents mentioned here and related literature as reported in the Reference section below.
Author Affiliation
Lorena Pereira and Milena Barrozo are co-founders and consultants at Vita Regulatory Affairs Consulting, a company based in Portugal and Brazil. Piero De Filippis is the StabilityHub EU Regional Contributor, based in Italy.
Comments and Information Requests
Do you have a request for information or clarification from the blog authors, or wish to share your experience in this area with the StabilityHub community? Please send an email to the authors at both info@vitaraconsulting.com and piero.df@outloook.com with “Comment on StabilityHub blog on FDS for ANVISA’ as the subject. Alternatively, you can use the contact tool in the StabilityHub web site, using the ‘Questions and Suggestions’ reason for contact.
The Blog authors will collect Questions & Answers and comments and respond in a document that will be posted in a future blog.
References (Part 3)
- ANVISA Resolução De Diretoria Colegiada [Collegiate Board of Directors Resolution] RDC – Nº 53. ‘Estabelece parâmetros para a notificação, identificação e qualificação de produtos de degradação em medicamentos com substâncias ativas sintéticas e semissintéticas, classificados como novos, genéricos e similares, e dá outras providências’ [‘Establishes parameters for the notification, identification and qualification of degradation products in medicines with synthetic and semi-synthetic active substances, classified as new, generic and similar, and takes other measures’] (4 December 2015 – in Portuguese) Direct link: http://antigo.anvisa.gov.br/documents/10181/3295768/%281%29RDC_53_2015_COMP.pdf/d38f507d-745c-4f6b-a0a6-bd250f2e9892 (last accessed 08 June 2021)
- ANVISA Guideline N° 04/2015 – Versão 1 ‘Guia para obtenção do perfil de degradação, e identificação e qualificação de produtos de degradação em medicamentos’ [‘Guide for obtaining the degradation profile, and identification and qualification of degradation products in medicines ’] (December 2015 – in Portuguese) – Direct link: http://antigo.anvisa.gov.br/documents/10181/2738062/Perfil+e+produtos+de+degrada%C3%A7%C3%A3o+em+medicamentos.pdf/c18a4857-9a5c-4292-a1bf-07af6cad6902 (last accessed 08 June 2021)
- ANVISA ‘Perguntas & Respostas – Assunto: RDC 53/2015 e Guia 4/2015’ [Questions & Answers – Subject: RDC 53/2015 and Guide 4/2015] Ed. 2.1. (October 2017 – in Portuguese) – Direct link https://www.gov.br/anvisa/pt-br/centraisdeconteudo/publicacoes/medicamentos/publicacoes-sobre-medicamentos/perguntas-e-respostas-rdc-53-2015-e-guia-04-2015.pdf (last accessed 08 June 2021)
- Lorena Pereira et al. ANVISA’s Guidance on Forced Degradation Studies Explained – Part 1. stabilityhub.com (Published on April 21, 2021)
- Lorena Pereira et al. ANVISA’s Guidance on Forced Degradation Studies Explained – Part 2. stabilityhub.com (Published on May 06, 2021)
- ANVISA Resolução de Diretoria Colegiada (Collegiate Board of Directors Resolution) RDC No. 318/2019. ‘Estabelece os critérios para a realização de Estudos de Estabilidade de insumos farmacêuticos ativos e medicamentos, exceto biológicos, e dá outras providências’ [‘Establishes the criteria for conducting Stability Studies for active pharmaceutical ingredients and drug products, except for biological, and provides other arrangements’] (Published in DOU No. 216, November 7, 2019 – in Portuguese) – direct link: https://www.in.gov.br/web/dou/-/resolucao-rdc-n-318-de-6-de-novembro-de-2019-226513805 (last accessed 08 June 2021)
- ICH Guideline Q1A(R2) ‘Stability Testing of New Drug Substances and Products’ (6 February 2003)
- ICH Guideline Q1B ‘Stability Testing: Photostability Testing of New Drug Substances and Products’ (6 November 1996)
- K.M. Alsante et al. The role of degradant profiling in active pharmaceutical ingredients and drug products. Adv. Drug Deliv. Rev. 59 (1):29-37 (2007)
- S.W. Baertschi et al. ‘Stress testing, a predictive tool’ in ‘Pharmaceutical stress testing – Predicting drug degradation’ 2nd Ed., pages 10-48. Informa Healthcare (2012)
- P. Tattersall et al. Impact from the Recent Issuance of ANVISA Resolution RDC 53/2015 on Pharmaceutical Small Molecule Forced Degradation Study Requirements. American Pharmaceutical Review. March 31 (2016)
- G. da Costa Nunes et al. Guidelines of forced degradation studies protocol and report of drug products according to RDC 53/2015 Infarma Ciências Farmacêuticas., 30 (3):194 202 (2018) (in Portuguese)
- ANVISA Resolução de Diretoria Colegiada (Collegiate Board of Directors Resolution) RDC No. 166/2017 ‘Dispõe sobre a validação de métodos analíticos e dá outras providências’ [Provides for the validation of analytical methods and other arrangements] (Published in DOU No. 141, July 25, 2017 – in Portuguese) – direct link: http://antigo.anvisa.gov.br/documents/10181/2721567/RDC_166_2017_COMP.pdf/d5fb92b3-6c6b-4130-8670-4e3263763401 (last accessed 08 June 2021)
- D.W. Reynolds et al. Available guidance and best practices for conducting forced degradation studies. Pharm. Tech. 26 (2):48-56 (2002)
- WHO Technical Report Series, No. 1010 – Annex 10 ‘Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products –Sect. 2.1.2 ‘Stress testing’ (2018)
- ICH Guideline M4Q(R1) ‘The Common Technical Document For the Registration of Pharmaceuticals For Human Use: Quality’ (12 September 2002)
- ANVISA Guideline N° 24/2019 ‘Guia para a Organização do Documento Técnico Comum (CTD) para o Registro e Pós-Registro de Medicamentos’ [Guide for the Organization of the Common Technical Document (CTD) for the Registration and Post-Registration of Medicines ] (14 August 2019) – Direct link https://pesquisa.anvisa.gov.br/upload/surveys/91575/files/Guia%20CTD.pdf (last accessed 08 June 2021)
- Whitepaper “Differences between CTD Brazil and EU: a look at CMC requirements”, Vita Regulatory Affairs Consulting Web Site at https://www.vitaraconsulting.com/news/2021/2/19/whitepaper-diffferences-between-ctd-brazil-and-ue-a-look-at-cmc-requirements (last accessed 08 June 2021)
Share This Article with the Stability Community!
February 28, 2026

Ben Was Right QC and analytical labs are facing heightened expectations as method lifecycle principles, data integrity, and digital lab tools reshape the regulatory [...]
February 6, 2026

If you oversee or participate in a medical product Stability program, you have been, or will be involved in an audit or inspection. While inspections [...]
January 11, 2026

In the tightly regulated world of pharmaceutical, medical device, and biologics manufacturing, contract storage facilities – warehouses or off-site stability storage facilities - play [...]
Share your questions and experiences
A stabilitarian encounters new situations every day. StabilityHub’s discussion forums give Stabilitarians an opportunity to ask questions and offer solutions to specific scenarios. Join in the conversations with other Stabilitiarians and share your knowledge!
A stabilitarian encounters new situations every day. StabilityHub’s discussion forums give Stabilitarians an opportunity to ask questions and offer solutions to specific scenarios. Join in the conversations with other Stabilitiarians and share your knowledge!