
Introduction
ANVISA, the Brazilian human health regulatory authority, has issued in the recent years a number of documents describing in detail what is expected to be presented on Forced Degradation Studies and how they should be designed and managed, and which relevant data should be evaluated (Ref. 1-3) .
Because of the increasing importance of the Brazilian pharmaceutical market in recent years, and the increasing acknowledgment of ANVISA as a leading world-wide Regulatory Authority, as discussed later in this blog, the authors thought that an article summarizing in an easy and schematic way, the key elements to consider when designing and assessing data from a FDS intended for ANVISA submission could be of interest and use to the StabilityHub community.
To help the reader better understand ANVISA FDS requirements, the first blog in this series will include high level and background information from ICH, WHO and ANVISA guidelines.
Stress testing studies (also known as Forced Degradation Studies (FDS), as they will be referred to in the present blog) are performed by exposing a drug substance (DS) and/or a drug product (DP) to exaggerated storage conditions (such as e.g., high temperatures, high humidity at solid state, wide pH range or highly oxidative conditions in solution, or high photo-exposure levels), to determine purposeful DS or DP degradation pathways to which they are sensitive (such as e.g., hydrolysis, oxidation or photolysis).
The first target of these studies, described at a high level in the stability guidelines Q1A(R2) and Q1B (Ref. 4&5) of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human
Use (ICH, i.e., formerly International Conference on Harmonization, before 2015), is to determine intrinsic stability and specific degradation pathways & products of a DS or a DP. The degradation products found in
stress studies are considered when developing and validating the stability-indicating analytical procedures used for the formal stability testing of the DS and DP.
Forced Degradation Studies according to ICH guidelines
A ‘stress testing’ condition (i.e., FDS) is one of the stability testing condition categories considered by the ICH Stability Guideline Q1A(R2), in addition to long-term and accelerated ones (Ref. 4a) .
Each one has a different target; the long-term condition to determine stability under the recommended storage conditions and the accelerated conditions to increase the rate of chemical degradation or physical change of a DS or DP respectively (Ref. 4a).
The stress testing target is ‘to elucidate the intrinsic stability of a drug substance’ (Ref.4a) and validate the stability-indicating power of the analytical method that will be used in the long-term and accelerated stability studies of a DS or a DP.
Stress testing “is normally carried out under more severe conditions than those used for accelerated testing” (Ref.1a) . The FDS conditions are detailed in the Q1A(R2) guideline for the drug substance and they “should include the effect of temperatures (in 10°C increments (e.g., 50°C, 60°C, etc.) above that for accelerated testing), humidity (e.g., 75% RH or greater) where appropriate, oxidation, and photolysis” as well as “…hydrolysis across a wide range of pH values when in solution or suspension.” (Ref.4b) .
Stress testing, according to ICH Q1A(R2) guideline “can help identify the likely degradation products, which can in turn help establish the degradation pathways and the intrinsic stability of the molecule and validate the stability indicating power of the analytical procedures used.” (Ref.4b) .
In other terms, the DS is exposed to the above-mentioned set of storage conditions, known to determine degradation (e.g., hydrolytic, oxidative, or photolytic) in sensitive compounds. This is done in order to assess both the compound susceptibility to degrade due to main atmospheric agents (i.e., temperature, moisture, oxygen and light) and the possible degradation routes and products. These degradation products will be considered when developing and validating the stability-indicating analytical procedures used for the formal stability testing of the active.
The final goal of a FDS is to support the development and validation of a stability-indicating analytical method by demonstrating for the DS its selectivity, considering its synthetic and degradation impurity contents, or for the DP, proving its capacity to detect all possible degradation products, separated from impurities of both the API and the excipients, allowing the evaluation the DS or DP stability during its retest period or shelf life, respectively.
While stress testing for the DS is quite well detailed in ICH Q1A (R2), no major detailed information is included for the DP, whose FDS are therefore left to common industrial best practices (Ref.6-8) . For submission in Brazil, a detailed regulation and guidelines for FDS in drug products are in force (Ref.1-3) . These will be explained in the next parts of this blog.
The ICH Q1A(R2) guideline explicitly indicates that FDS results “will form an integral part of the information provided to regulatory authorities” (Ref.4b).
The inclusion of FDS data in regulatory registration documents for marketing based on the Common Technical Document (CTD) format, is also an explicit expectation in the ICH M4Q(R1) guideline on CTD (Ref. 9 . FDS data should be included in the DS Stability section No. 3.2.S.7 of the application, both as summary and tabulated data.
FDS recommendations in the World Health Organization (WHO) stability guideline (covering countries world-wide) (Ref.10) are similar to those in ICH Q1A (R2), with a few minor differences. The most important additional information is the recommendation for the level of degradation to be obtained during an FDS. According to the WHO guideline (Ref. 10a) degradation should typically occur to a small extent (typically 10-30%) or, in total absence of degradation products, expose the product up to 10-days storage under the particular stress condition. The minimum level of degradation recommended by the WHO to be achieved in a FDS is aligned to the one adopted by ANVISA in the internal regulation, RDC 53/2015 (Ref. 1) , (i.e., more than 10%). Regulation RDC 53/2015 will be discussed in the next parts of this blog.
ANVISA, the Brazilian Health Authority
The ANVISA (Agência Nacional de Vigilância Sanitária) is the National Health Surveillance Agency in Brazil.
It is a relatively young Regulatory Authority created in 1999 by law 9.782/1999, and is characterized by its administrative independence, financial autonomy, and the stability of its board of directors (Ref. 11) . In the Brazilian federal public regulatory structure, the agency is connected to the Ministry of Health.
ANVISA is ruled by a Collegiate Board of Directors made up of five members, and has the duties of health surveillance, including powers to regulate and to ensure the health and safety of consumers.
ANVISA´s main duties, as defined by law 9.782/1999, are to::
- Coordinate the Brazilian Health Surveillance System
- Establish Rules
- Propose regulations, guidelines, and actions
- Issue authorization for companies’ activities
- Issue Marketing Authorization for products
- Issue licenses for import and export of products
- Issue infraction notices and application of penalties
- Carry out toxicological and pharmacological surveillance
It is important to mention that the Brazilian twenty-six federated states, including one federal district and municipal governments, also have sanitary surveillance bodies, whose competence involves inspection and licensing of facilities, as well as local surveillance of health products and compliance with health legislation.
The products under ANVISA´s control are medicines for human use, biological products, radiopharmaceuticals, medical devices, disinfectants, blood products, diagnostic materials, cosmetics, food and tobacco. Additionally, any product that may carry the possibility of risk to human health, obtained by genetic engineering, or by any other procedure, or submitted to radiation sources that may result in potential risk to human health is also under ANVISA control.
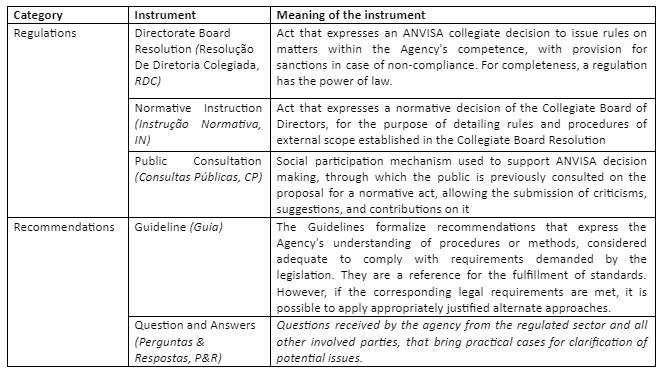
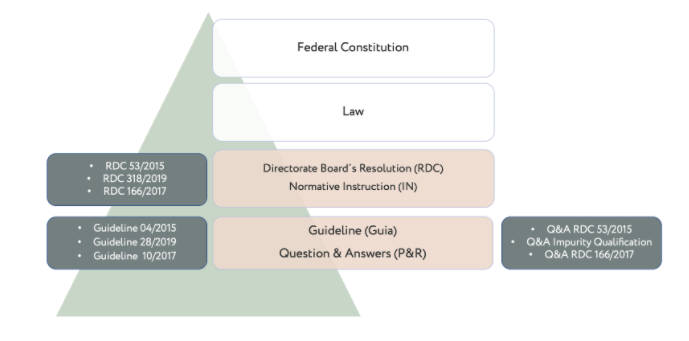
The most common instruments used by ANVISA to exercise its role of regulator (i.e., proposing regulations, guidelines, and actions for health surveillance), are described in its internal statute defined per RDC nº 255/2018, and are detailed in the table below.
In the preceding years, ANVISA has gone through an evolutionary process, which began in 2012, with ANVISA getting closer to ICH.
in December 2015, after a period of many changes and internal restructuring, the agency was accepted as an observer member of the ICH. In November 2016, it became a regulatory member of ICH. It is important to mention that to this date, ANVISA is the only Latin American Regulatory Authority in this position.
In November 2019, ANVISA became a member of the ICH management committee with the Regulatory Authorities of the United States, Canada, European Union, Japan, Switzerland, China, Singapore, and Republic of Korea. This position is fostering an international harmonization of requirements, ensuring that the drug products approved by ANVISA have the same quality as the products approved in the main international markets.
In addition, in November 2020, ANVISA was approved for membership of the PIC/S (Pharmaceutical Inspection Co-operation Scheme), becoming active in January 2021, having achieved international recognition as an agency with high technical rigor in the evaluation of drug products and for excellence of inspections in Good Manufacturing Practices (GMP) of drug products and drug substances for human use.
Acknowledgements
The authors wish to thank Deborah Fernandes, from Vita Regulatory Affairs Consulting, for her highly appreciated support in the preparation of Part 1 of this blog.
Do you have a request for information or clarification from the blog authors, or wish to share your experience in this area with the StabilityHub community? Please send an email to the authors at both info@vitaraconsulting.com and piero.df@outloook.com with “Comment on StabilityHub blog on FDS for ANVISA’ as the subject. The Blog authors will collect Questions & Answers and comments and respond in a document that will be posted in a future blog.
References (Part 1)
- ANVISA Resolução De Diretoria Colegiada (Collegiate Board of Directors Resolution) RDC – Nº 53. ‘Estabelece parâmetros para a notificação, identificação e qualificação de produtos de degradação em medicamentos com substâncias ativas sintéticas e semissintéticas, classificados como novos, genéricos e similares, e dá outras providências’ [Establishes parameters for the notification, identification and qualification of degradation products in medicines with synthetic and semi-synthetic active substances, classified as new, generic and similar, and takes other measures] (4 December 2015 – in Portuguese) Direct link: http://antigo.anvisa.gov.br/documents/10181/3295768/%281%29RDC_53_2015_COMP.pdf/d38f507d-745c-4f6b-a0a6-bd250f2e9892 (last accessed 08 April 2021)
- ANVISA Guia N° 04/2015 – Versão 1 ‘Guia para obtenção do perfil de degradação, e identificação e qualificação de produtos de degradação em medicamentos’ [‘Guide for obtaining the degradation profile, and identification and qualification of degradation products in medicines ’] (December 2015 – in Portuguese) – Direct link: http://antigo.anvisa.gov.br/documents/10181/2738062/Perfil+e+produtos+de+degrada%C3%A7%C3%A3o+em+medicamentos.pdf/c18a4857-9a5c-4292-a1bf-07af6cad6902 (last accessed 08 April 2021)
- ANVISA ‘Perguntas & Respostas – Assunto: RDC 53/2015 e Guia 4/2015’ [Questions & Answers – Subject: RDC 53/2015 and Guide 4/2015] Ed. 2.1. (October 2017 – in Portuguese) – Direct link https://www.gov.br/anvisa/pt-br/centraisdeconteudo/publicacoes/medicamentos/publicacoes-sobre-medicamentos/perguntas-e-respostas-rdc-53-2015-e-guia-04-2015.pdf/@@download/file/Perguntas%20e%20Respostas%20-%20RDC%2053%202015%20e%20Guia%2004%202015.pdf (last accessed 08 April 2021)
- International Council for Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Guideline Q1A(R2) ‘Stability Testing of New Drug Substances and Products’ – a) Glossary – b) Sect. 2.1.2. Stress Testing (6 February 2003)
- ICH Guideline Q1B ‘Stability Testing: Photostability Testing of New Drug Substances and Products’ (6 November 1996)
- Dan W. Reynolds et al. Available guidance and best practices for conducting forced degradation studies. Pharm. Tech. 26 (2):48-56 (2002)
- Karen M. Alsante et al. The role of degradant profiling in active pharmaceutical ingredients and drug products. Adv. Drug Deliv. Rev. 59 (1):29-37 (2007)
- Ragine Maheswaran. FDA Perspectives: Scientific Considerations of Forced Degradation Studies in ANDA Submissions. Pharm. Techn. 36 (5):73-80 (2012)
- ICH Guideline M4Q(R1) ‘The Common Technical Document for The Registration of Pharmaceuticals for Human Use: Quality’ (2002)
- WHO Technical Report Series, No. 1010 – Annex 10 ‘Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products – a) Sect. 2.1.2 ‘Stress testing’ (2018)
- Kim Huynh-Ba et al. ANVISA: an introduction to a new regulatory agency with many challenges. AAPS Open 4:9 (2018)
Share This Article with the Stability Community!
February 28, 2026

Ben Was Right QC and analytical labs are facing heightened expectations as method lifecycle principles, data integrity, and digital lab tools reshape the regulatory [...]
February 6, 2026

If you oversee or participate in a medical product Stability program, you have been, or will be involved in an audit or inspection. While inspections [...]
January 11, 2026

In the tightly regulated world of pharmaceutical, medical device, and biologics manufacturing, contract storage facilities – warehouses or off-site stability storage facilities - play [...]
Share your questions and experiences
A stabilitarian encounters new situations every day. StabilityHub’s discussion forums give Stabilitarians an opportunity to ask questions and offer solutions to specific scenarios. Join in the conversations with other Stabilitiarians and share your knowledge!
A stabilitarian encounters new situations every day. StabilityHub’s discussion forums give Stabilitarians an opportunity to ask questions and offer solutions to specific scenarios. Join in the conversations with other Stabilitiarians and share your knowledge!