
This blog post is Part 2 of the series and comes to us from Piero De Filippis, StabilityHub EU Regional Contributor, Verona (Italy).
Introduction
This blog reports key points to consider when assigning or extending a retest period (RP) or a shelf-life (SL) to a drug substance (DS, i.e. an API) or to the related Investigational Medicinal Product (IMP), with relevant label storage conditions, to support clinical studies in the European Union (EU); it includes both an EU regulatory perspective and some notes from the author’s experience. This is part 2 of a 2-part blog: the first part covered the IMP stability study design for EU (Ref.1).
Stability data reporting in the IMPD
The available stability data should be presented in a tabulated form for both the DS and the relevant IMP in the Investigational Medicinal Product Dossier (IMPD) (Ref.2a&b), the regulatory document including all the Chemistry, Manufacturing and Control information on the IMP. As mentioned in part 1 of this blog , the approval of the IMPD by the EU Member state regulatory authority where the clinical trial will be run, is a prerequisite for the clinical study start.
For IMP’s for Phase 1 clinical trials, the EMA IMPD guideline would allow to progress the IMPD with a commitment that “an ongoing stability program will be carried out with the relevant batch(es) and that, prior to the start of the clinical trial, at least studies under accelerated and long-term storage conditions will have been initiated. (…) Supportive data from development studies should be summarised in a tabular overview.”. As a note, in general terms it is not within the author’s experience to file an IMPD in the EU to support a Phase I clinical trial without any minimum (e.g. 1 month) representative IMP stability data, the EU regulators in the author’s view being generally keen to see at least some stability data on the drug substance and the IMP itself (or a strong supporting rationale and/or data).
For drug substances covered by a pharmacopoeial monograph, confirmation that DS batches used in IMP preparation will meet specifications at the time of use in manufacturing is also considered acceptable by the EMA IMPD guideline (Ref.2a).
Assignment of a retest period and shelf-life and relevant storage conditions to drug substances and IMPs
The definition and inclusion in the IMPD of a retest period (RP) for the DS or a shelf-life (SL) for the IMP is an explicit requirement for EU (Ref.2a&b).
While for the DS there is no detailed indication in the EMA IMPD guideline on how to derive a RP from available stability data (Ref.2a), some detailed rules are reported in this guideline to derive a SL assignment to the IMP on the basis of available stability data (Ref.2b).
For the SL assignment to an IMP, the EMA IMPD guideline allows a “fourfold extrapolation of accelerated stability data may be acceptable up to a shelf life of 12 months and an extrapolation of + max 12 months to long-term stability data available (at least 6-months) may be acceptable for a shelf life of more than 12 months”. “…other schemes may be possible but should be justified” (Ref.2b).
Table 1 below illustrates in practice how to derive a SL for an IMP on the basis of available stability data, according to the EMA IMPD guideline rules (Ref.2b).
Table 1 – Example of maximum assignable shelf-lives to IMP’s according to EMA IMPD guideline criteria.
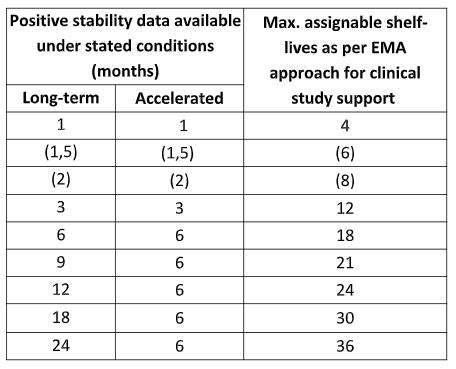
As it can be seen, these EMA IMP rules allow a wider SL assignment than the ones in the ICH guideline Q1E (Ref.3) mainly in the earlier time points. E.g.: an 18 months SL can be assigned on the basis of 6 month data to an IMP according to EMA IMP criteria, vs. 12 months SL on the basis of the same stability study length according to ICH Q1E criteria for a stable product to be stored at room temperature.
Explicitly highlighted terms of applicability of the abovementioned rules by the EMA IMPD guideline are that “Results at long-term as well as at accelerated storage conditions are available” and that “ No significant changes in stability behaviour are observed. If any observed, justification should be provided”
In case of instabilities or variabilities, it may be appropriate to perform and assess a trend analysis of data with any statistical support and/or to apply the ICH Q1E rules, as applicable, to assign a RP or a SL to the DS or to the IMP.
As mentioned above, a scheme for an analogue RP assignment to DS is not reported in the DS stability section in the EMA IMPD guideline (Ref.2a). However, in author’s experience it may be convenient, whenever possible, to apply the same assignment and extension scheme to both DS and IMPs, for ease of regulatory approval and internal consistency of approach.
The rules to be applied to define labeling storage conditions and restrictions are not reported in the EMA IMPD guideline but in the author’s view, it may be convenient to derive them from the EMA guideline for storage labeling for market (Ref. 4). However, it is always appropriate to establish a labeled storage condition for an IMP, even when the above guideline would allow not to do so (i.e. for very stable drug products).
As mentioned in Part 1 of this blog, when appropriate, an in-use SL should be also assigned to some IMPs when applicable (e.g., to non-oral solid preparations intended to be administered after reconstitution, dilution or mixing and for products in multi-dose containers) (Ref.2b).
In the EU there is an explicit regulatory expectation that a drug product on the market should be transported at the approved storage conditions (Ref.5&6). Therefore, for whatever reason, if the IMP planned transport condition differs from the proposed storage conditions, it is author’s view that this should be detailed in the IMPD with relevant supporting data to get regulatory approval for this difference.
Retest period or shelf-life extension
In general, the extension of the RP for the DS or of the SL for the IMP in support of a clinical study requires in the EU, the submission of the updated stability data to the competent regulatory authority within a substantial amendment document. This is approved by the regulatory authority before possibly extending clinical supplies expiry dates. This approach could, however, easily cause delays in clinical study support when pursuing continuous shelf-life extensions to support the use of IMP lots in an ongoing clinical study.
It is however possible in the EU, as reported in the EMA IMPD guideline (Ref. 2c), to extend RP and SL on the basis of updated available stability data without submitting new data in a substantial modification document to the authority, by clearly indicating in the IMPD and receiving approval of the criteria the applicant will apply to extend the RP or SL or the storage conditions.
It is obviously highly important that the planned extension criteria are very clearly stated and detailed in the IMPD to obtain approval of this route by the regulatory authority. This detail should give the Authority the confidence that the applicant will extend the RP/SL on a sound basis without a prior regulatory review of stability data.
The EMA Q&A page in Part 2 (Ref. 7) reports additional information on this topic. In the details, the information reported in the IMPD should include an extension protocol limiting the maximum time period for extrapolation. Additionally, in case of significant trends in stability data observed during long-term and accelerated testing, the sponsor should commit to notify authorities of any shelf-life extension as a substantial amendment.
Overall Conclusions
Although the main ICH stability guidelines are explicitly not intended to be applied to clinical phase, stability testing study design & data evaluation to support a clinical study is currently highly influenced by the main ICH stability guidelines (such as e.g. ICH Q1A, Q1B and Q1E) as applicable with the proper modifications required by the phase of the development.
However, when designing a stability study and evaluating stability data to support a clinical study in the EU, a number of local expectations (as e.g. defining and getting approval for a shelf-life to the IMP) and opportunities (as e.g. possible application of an initial shelf-life assignment and following extension scheme wider than that of ICH Q1E and applying an automatic shelf-life extensions plan on the basis of agreed criteria with regulatory authorities), as reported in the EMA IMPD guideline, should be considered.
Do you have any information or a request for clarification for this blog author or any experience in this area that you wish to share with the StabilityHub community? Please send your question or comment to the author at piero.df@outlook.com with the subject ‘Comment on blog on stability study for an IMP in EU – Part 2.’ Or you may click the button below. Questions & Answers and comments will be collected by this blog author in a document that will be posted after this blog.
Reference (Part 2)
- P. De Filippis. Stability Testing Study Design and Data Evaluation to Support a Clinical Study in the European Union – Part 1: Stability Study Design for an Investigational Medicinal Product. StabilityHub (online 02 March 2021)
- European Medicines Agency (EMA) guideline EMA/CHMP/QWP/545525/2017. Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials’ – a) Sect. 2.2.1.S.7 ‘Stability’; b) Sect 2.2.1.P.8 ‘Stability’; c) Sect. 9. ‘Changes to the investigational medicinal product and auxiliary medicinal product with a need to request a substantial modification to the IMPD’ and Tables 1 and 3 [Link: https://www.ema.europa.eu/en/requirements-chemical-pharmaceutical-quality-documentation-concerning-investigational-medicinal – last accessed 24 February 2021]
- International Council for Harmonisation (ICH) guideline Q1E. Evaluation of Stability Data (August 2003) [link https://www.ema.europa.eu/en/ich-q1e-evaluation-stability-data last accessed 24 February 2021]
- European Medicines Agency (EMA) guideline CPMP/QWP/609/96/Rev 2. Guideline on declaration of storage conditions: A: in the product information of medicinal products – B: for active substances. (19 November 2007) [Link: https://www.ema.europa.eu/en/declaration-storage-conditions-medicinal-products-particulars-active-substances-annex – last accessed 24 February 2021]
- European Commission Guideline 2013/C 343/01 – Good Distribution Practice of medicinal products for human use – Sect. 9.2. Transportation (5 November 2013) [Link: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:C:2013:343:0001:0014:EN:PDF – last accessed 24 February 2021]
- European Commission Good Distribution Practice For Medicinal Products For Human Use , Questions and Answers – Version 1.0 – Question 22 (28 March2014) [Link: https://ec.europa.eu/health//sites/health/files/files/gdp/2014-04_qas_.pdf – last accessed 24 February 2021]
- European Medicines Agency Quality of medicines questions and answers: Part 2. Specific types of product – Quality of investigational medicinal products – Question 3. Shelf-life extension [link: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/qa-quality/quality-medicines-questions-answers-part-2 – last accessed 24 February 2021]
Share This Article with the Stability Community!
February 28, 2026

Ben Was Right QC and analytical labs are facing heightened expectations as method lifecycle principles, data integrity, and digital lab tools reshape the regulatory [...]
February 6, 2026

If you oversee or participate in a medical product Stability program, you have been, or will be involved in an audit or inspection. While inspections [...]
January 11, 2026

In the tightly regulated world of pharmaceutical, medical device, and biologics manufacturing, contract storage facilities – warehouses or off-site stability storage facilities - play [...]
Share your questions and experiences
A stabilitarian encounters new situations every day. StabilityHub’s discussion forums give Stabilitarians an opportunity to ask questions and offer solutions to specific scenarios. Join in the conversations with other Stabilitiarians and share your knowledge!
A stabilitarian encounters new situations every day. StabilityHub’s discussion forums give Stabilitarians an opportunity to ask questions and offer solutions to specific scenarios. Join in the conversations with other Stabilitiarians and share your knowledge!